登录
记住用户名密码
记住用户名密码
以清洁能源H2为燃料的质子交换膜燃料电池应用前景广阔,而经蒸汽重整、水煤气变换等制备H2残留的微量CO将严重毒化Pt电极。富氢氛围中CO优先氧化(PROX)是进气路上在线去除痕量CO的理想方式,负载于过渡金属氧化物的高分散Pt体系被认为是极具前景的PROX催化剂。
此前,华中科技大学单斌教授和陈蓉教授团队(微纳材料设计与制造研究中心)基于选择性ALD方法制备了Pt单原子镶嵌于CoOx团簇的精细纳米结构,经H2预还原处理后表现出优异的CO氧化活性和选择性(https://doi.org/10.1039/D0TA01151G),但CO与H2的竞争氧化机理和H2处理的影响机制尚不明晰。
基于此,课题组进一步采用第一性原理结合“态到态”微反应动力学方法系统对比研究了有无H2处理时CoOx负载Pt单原子(Pt1/CoOx)体系的CO PROX反应机理,揭示了H2还原处理对PROX性能的微观促进机制。相关工作以“Promotional Effect of H2 Pretreatment on the CO PROX Performance of Pt1/Co3O4: A First-Principles-Based Microkinetic Analysis”为题,发表于《ACS Applied Materials & Interfaces》(https://doi.org/10.1021/acsami.2c00775)。博士研究生刘璋为论文的第一作者,单斌教授和陈蓉教授是共同通讯作者。
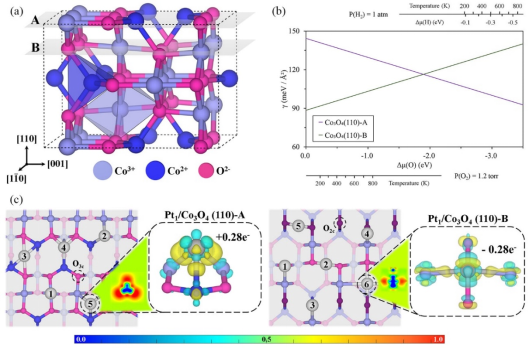
本论文首先根据TEM和O化学势计算的结果,分别以Co3O4 (110)富Co的type A面封端和富O的type B面封端作为有无H2处理时负载Pt单原子的载体表面,如图1所示,并通过结合能测试建立稳定的Pt1/Co3O4(110)-A(B)构型。
对于无H2处理的Pt1/Co3O4(110)-B体系,建立如图2示意的反应网络,CO和H2竞争氧化的本质体现于二者对表面活性O原子的竞争反应。DFT计算结果表明,CO相较于O2对Pt1位点的竞争吸附占据绝对优势,且贫Co的B面依靠Pt-Co位点分解O2的势垒较高。

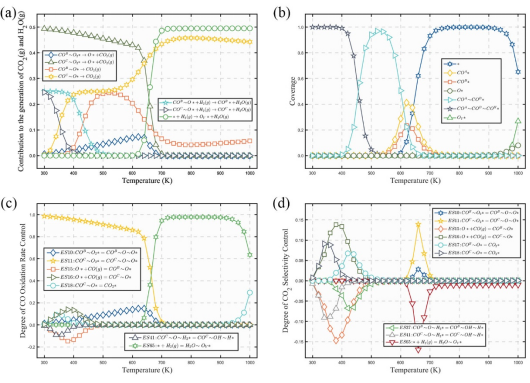
为进一步定量评估不同温度下的催化性能,根据单原子模型活性位点限域的特点,将Pt1及其邻域组成的复合位点予以整体化处理,局域化学环境不同的中间(共吸附)物种作为独立的反应态,将基元反应视为相邻反应态间的跳跃,建立“态到态”(state-to-state, STS)微反应动力学模型。求解稳态方程,如图3所示,结果表明,CO的(共)吸附态在在较宽的低温区间覆盖了表面、毒化了Pt位点,导致较差的低温活性和选择性,且高势垒的O2分解是低温区间的决速步骤。温度较高时,H2(g)更易直接结合表面氧空位产生H2O(g),导致CO2选择性降低。
对于无H2处理的Pt1H/Co3O4(110)-A体系,建立如图4示意的反应网络,H2处理能够暴露富Co面特有Co~Co二聚体位点,其具有吸附、分解O2能力强的优势,而CO依然强吸附于Pt单原子,二者共同构成双功能位点、遵循非竞争Langmuir-Hinshelwood(LH)机理。此外,表面活性H能够扭动O2辅助其分解,并通过生成COOH物种完成较低势垒的CO氧化。

同理,建立并求解关于Pt1H/Co3O4(110)-A体系的STS微反应动力学模型,如图5所示。结果表明,CO的(共)吸附态不再是低温区间的主要覆盖物种,表明其毒化效应得以消除。此外,H辅助路径主要贡献了CO2(g)的产生,表明其对CO氧化活性的促进作用。由于H2O(g)的产生主要来源于H2(g)对O2直接解离路径的进攻,表明H辅助路径同样能够促进CO2低温选择性。